Selection of target sites. Since the maize genome (2.7 Gb) has expanded in size because of the recent amplification of eight major retrotransposon families (SanMiguel and Bennetzen, Ann. Bot. 81:27-44, 1998; Meyers et al., Genome Res. 11:1660-76, 2001), a certain length of genomic DNA has to be sequenced to facilitate a comparison between the larger maize and the smaller sorghum genome (0.77 Gb). In the case of sorghum, usually we select a single bacterial artificial chromosome (BAC) clone for each locus. These BACs contained inserts that ranged between 80 and 160-kb. These clones were chosen using maize sequences of a number of selected loci. After sequencing the sorghum clones and subjecting them to gene prediction programs, sorghum sequences were used to select maize BAC clones. Such reverse screening gave us an indication whether orthologous gene sequences are present and closely linked. However, sometimes translocation of paralogous sequences could yield two BAC clones from different chromosomal locations.
A total of 15 maize loci have been used as entry points into the genome. Twelve of these loci represent six duplicate factors (orthologous positions) in the maize genome (Rhoades, Am. Nat. 85:105-110, 1951). The remaining three loci do not represent duplicate factors, but are special gene clusters.
Two examples involve two closely linked orthologous regions. Two BAC clones, Z477F24 and Z178A11, are from chromosome arm 1L and contain the tbp1 (TATA box-binding protein) and the tb1 (teosinte-branched) locus at position 190.8 and 197.6, respectively; genetic positions have been taken from the Pioneer composite map, 1999, which has hyperlinks to all references (see MaizeDB). This segment is duplicated on chromosome arm 5S around position 55. Two BAC clones, Z474J15 and Z195D10, contain the tbp2 and tb2 loci, respectively. We have already sequenced the orthologous clones from sorghum. SB32H17 contains the TATA box-binding protein gene. Sorghum clone SB45I19 contains the ortholog to maize tb2 on maize chromosome arm 5S, rather than tb1. Another predicted gene linked to tb2 is present in sorghum, but not maize 1L. It will be interesting to see what function the orthologous genes on 5S might have. In the case of tb2, we already know that it is degenerate and probably not expressed, which may explain why tb1 acts as a single Mendelian factor in crosses with teosinte (J. Doebley, pers. comm.). Interestingly, tb1 appears to in a gene-poor region, as it is the only gene that we can find with certainty within 130-kb, while tb2 is contained within a gene-rich region, indicating the divergence of orthologous regions in the maize genome because of retrotranspositions.
Other closely linked duplicate factors are orange pericarp (orp) and fertilization-independent-endosperm (mfie), mapping about 1 cM apart on 4S and 10S. We are currently sequencing Z332A24 and Z409L08, which contain the genes for orp1 (4S, 66.3) and orp2 (10S, 61). The orthologous clone SB18C08 of sorghum has already been sequenced. The orp1 and orp2 loci encode a tryptophan synthase b subunit that is highly conserved from Arabidopsis to rice. Z273B07 (mfie1) and Z078P04 (mfie2) contain the other duplicate factors that are closely linked to the Orp genes (Lai and Messing, in preparation). In Arabidopsis, the FIE gene is important for seed development and is subject to parental imprinting (Luo et al., Proc. Natl. Acad. Sci. USA 97:10637-10642, 2000). In maize, only one of the duplicate factors undergoes imprinting. Since, in mammalian systems, genomic imprinting may involve chromosomal regions that contain several genes (Leighton et al., Nature 375, 34-39, 1995), comparison of the two subgenomes of maize in this region may provide new insights into parental imprinting in plants.
The fifth set of duplicate markers provides an example of regions within two maize chromosomes that have arisen from an ancestral chromosome by a large chromosomal inversion (Devos et al., Genetics 138:1287-1292). RFLP mapping supports this hypothesis because of the reverse order of collinear markers. We are currently sequencing two BAC clones Z438D03 and Z576C20 that contain the c1 (chromosome 9) and the pl1 (chromosome 6) loci, respectively, which are positioned in the center of this inversion (c1, 33.1 on 9; pl1, 75.4). We have also sequenced an orthologous clone from sorghum, SB35P03, to investigate which subgenome is closer to sorghum. The C1 and Pl1 genes encode myb-like DNA binding proteins that control pigment synthesis in maize; a closely conserved sequence in rice is located on rice chromosome 2. The duplicate factors in maize differ in their tissue specificity, indicating that gene regulation rather than gene function has diverged. Another duplicate factor that represents transcription factors are r1 (chromosome 10L, 105.3) and b1 (chromosome 2S, 65.6). A contig has been formed from two overlapping BAC clones, Z138B04 and Z333J11, from the r1 region. Z092E12 represents the b1 locus. The orthologous region from sorghum is on SB20O07. These two loci have been intensively studied because of imprinting and paramutation.
The last three loci are not based on duplicate factors, but on clusters of related genes. One example involves resistance against Puccinia sorghi or maize rust. There are several loci, rp1, rp5, rp6, and rpp9 (resistance to Puccinia polysora and sorghi) that map to chromosome arm 10S from 8.5 to 10.1. It has been estimated that 11 or more genes are spread over 400 kb (Ramakrishna and Bennetzen, in preparation). We have sequenced two BACs from this interval from a different B73 source. The two BACs contain a 43-kb direct repeat that contains both rp1 genes and retrotransposons, indicating that amplification of this region occurred in large blocks (Ramakrishna and Bennetzen, in preparation). The orthologous sequence in sorghum differs significantly. We have sequenced SB98N08 and SB95A23, which overlap. This sorghum contig seems to contain most of the orthologous rp1 genes. The rp1 genes are all very closely placed in sorghum and other gene sequences were also duplicated, but without the intervening retroelements present in maize. The second region contains members of the DIMBOA pathway. The genes in question are all involved in the biosynthesis of DIMBOA, which inhibits fungal, bacterial and insect damage to aerial parts and roots in Bx1 plants. Several genes, bx1, bx2, bx3, bx5, two cytochrome P450 loci and rp4 have been mapped to the interval between 13.1 and 16.9 on maize chromosome arm 4S. Again we have isolated orthologous clones from maize and sorghum, but we do not know yet whether the same compactness of the rp1 genes exists for these genes in sorghum as well. The third region contains a cluster of storage protein genes on chromosome 4S. This example required the construction of a new BAC library of BSSS53 because of our interest in the positional cloning of dzr1 (Chaudhuri and Messing, Proc. Natl. Acad. Sci. 91:4867-4871, 1994). Two overlapping clones contain 346-kb of maize genomic sequence (Song et al., Genome Res. 11:1817-1825, 2001). This region contains 22 tandem repeated 22-kDa a zein genes that are linked to the php200725 marker (27.3). Using this marker, orthologous clones have been isolated and sequenced from sorghum and rice (Song, Llaca, and Messing, in preparation).
Table 1 summarizes the current status of our BAC clones. All of the sequences under the heading �Finished� are being annotated and will be deposited into GenBank. Clones under the heading �In Production� are either at Phase I or Phase II assembly.
Table 1.
| Marker | Name | Map | Finished | Size bp | In Production |
| rp1 | rust | 8.5 � 10.1 C10 | Z163K15 | 95,078 | |
| resistance | Z238E11 | 99,156 | |||
| SB98N08 | 239,785 | ||||
| SB95A23 | 97,616 | ||||
| r1/b1 | red color | 65.6 C2 | Z092E12 | 147,198 | |
| booster | 105.3 C10 | Z138B04 | 115,734 | ||
| Z333J11 (B04 Ext.) | |||||
| SB20O07 | |||||
| tb1/2 | teosinte | SB45I19 | 77,947 | ||
| branched | 197.6 C1 | Z178A11 | 130,843 | ||
| 55 C5 | Z195D10 | 141,939 | |||
| TBD (A11 5� Ext.) | |||||
| orp1/2 | orange | SB18C08 | 159,669 | ||
| pericarp | 66.3 C4 | Z332A24 | |||
| 61 C10 | Z409L08 | ||||
| tbp1/2 | TATA-box | SB32H17 | 100,707 | ||
| binding | 190.8 C1 | Z477F24 | |||
| protein | 55 C5 | Z474J15 | |||
| c1/pl1 | colored | SB35P03 | 144,120 | ||
| aleurone | 33.1 C9 | Z438D03 | |||
| purple | 75.4 C6 | Z576C20 | |||
| plant | |||||
| bx3 | benzoxazin | S106M24 | |||
| 13.1 � 16.9 C4 | TBD | ||||
| mfie1/2 | fertilization | C4 | Z273B07 | ||
| independent | C10 | Z078P04 | |||
| endosperm
development |
|||||
| php200725 | RFLP marker linked to
22-kDa |
27.3 C4 | ZMRS204
ZMRS171 ZMRS072 |
435,084 | |
| zein/kafirin cluster | SB25M18
SB40L16 SB126P21 SB234M12 |
425,898 | |||
| 97.4 C11 | L01H19
T16F19 |
77,605
70,311 |
Bioinformatics. Because of the large size and number of BAC clones that were sequenced during this project we found the existing annotation tools to be inadequate for our purposes. Therefore, development of new computational tools for local and global alignments, along with semi-automation of existing methods of gene finding and annotations was undertaken.
Initially we leveraged the existing resources available on the web, FGENESH (www.softberry.com), GenScan (CCR-081.mit.edu/GENSCAN.html), BlastP and BlastN (www.ncbi.nlm. nih.gov), in combination with custom Perl scripts for sequence analyses. After analyses the results were manually reviewed, and a quality annotation of the BAC sequence data was produced. This annotated sequence was produced from reviewed data contained in an Excel spreadsheet. A Perl script converted the spreadsheet data into a graphic JPEG file. At present the descriptive fields for each gene are added manually, though we are working to also automate this final step. Additional work to integrate these processes in an easy-to-use web-based format is underway.
Besides combining existing gene finding methods into an integrated process, new methods of DNA sequence analyses are also being developed. Currently, a new method for comparing two DNA sequences for conserved regions while taking into account events like local inversions has been developed (do Lago et al. in preparation). While the details of this new method are beyond the scope of this report, a brief explanation is possible.
Most pairwise alignment programs use the paradigm of the Longest Common Subsequence (LCS). That is, they try to maximize the overall (global) alignment, but end up sacrificing the best local alignments in their search for the longest single sequence common to both sequences. Our method does not make this sacrifice and identifies local inversions.
Steps in the Alignment Process. 1) Original sequences are decomposed into fragments of equal length that may overlap. 2) Fragments from one sequence are aligned to fragments of the other sequence, based on their alignment scores. 3) Clustering is performed on the obtained assignment of fragments.
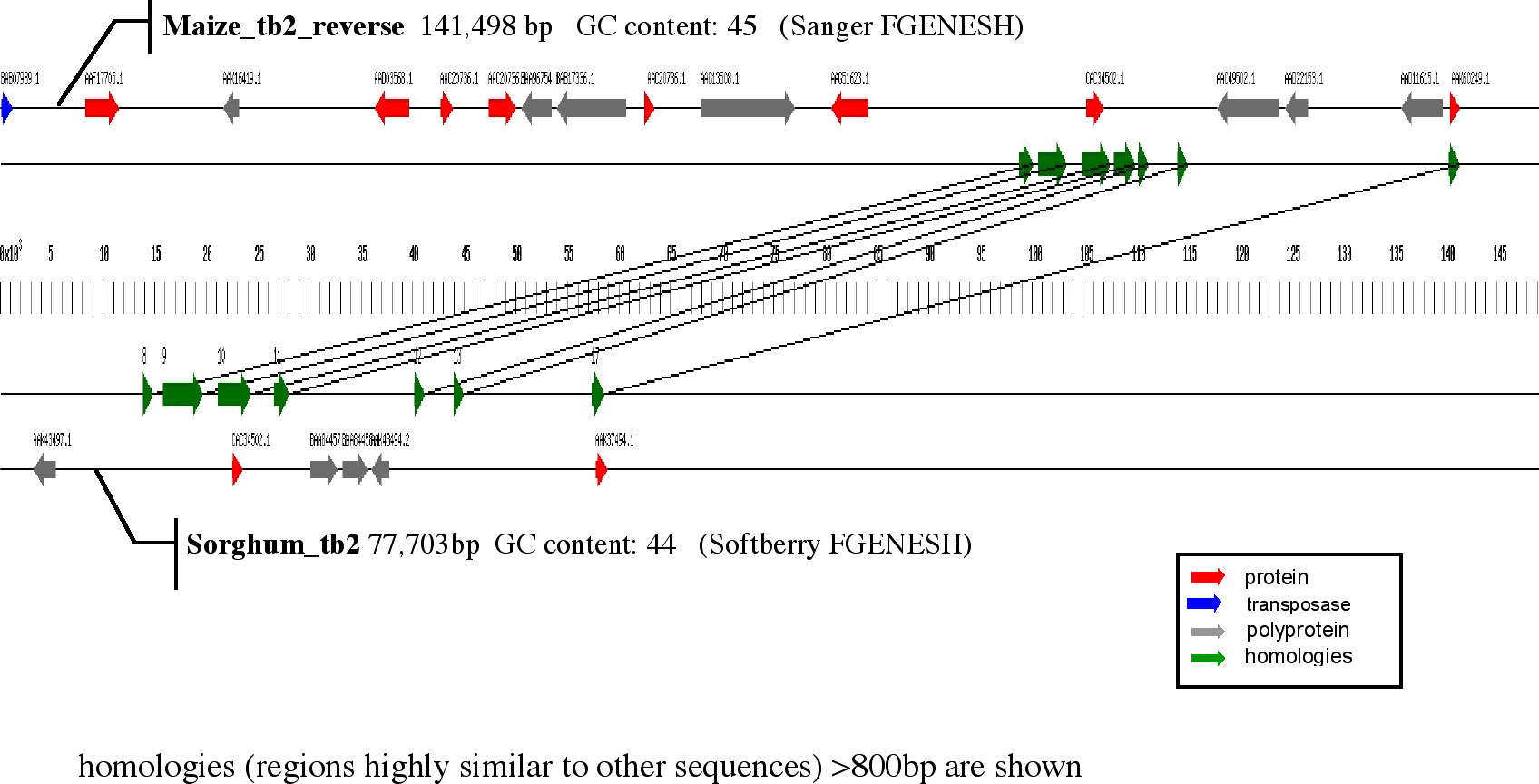
An example of the integration of gene prediction and global alignment is shown in Fig. 1. In this example, clone Z195D10 (maize tb2) and clone SB45I19 (sorghum tb2) are first subjected to gene prediction analysis and homology searches of predicted proteins with the respective databases. The results are presented in two horizontal bars. In addition, sequences are compared by the global alignment methods described above. The results are presented in a collinear fashion with the gene prediction results. The method is more sensitive since it does not rely on ORFs. Interestingly, regions of orthology can be extended beyond gene sequences.
Figure
1. Gene prediction analysis of the maize and sorghum tb2 regions.
Each sequence is first subjected to a gene prediction program as indicated.
The predicted proteins are then subjected to BLASTP. The graphic output
indicates the polarity of the predicted coding sequence with the accession
number of the best hit above it. In addition, each sequence is compared
by the new global alignment tool (do Lago et al., in preparation). Arrows
on both sides of the bp scale indicate conserved sequences and their polarity.
Conserved sequences between the two clones are indicated by the vertical
lines.
Return to the MNL 76 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}