Pyrosequencing is a new method of obtaining short segments of DNA sequence, typically up to 20 nucleotides, simultaneously on 96 different templates. Once the templates have been prepared, 96 templates can be sequenced in 15 minutes. The methodology was developed by P. Nyren (Nyrén, P., Analytical Biochemistry 167:235, 1987; Ronaghi, M. et al, Science 281:363, 1998). It relies on the stepwise elongation of the primer upon sequential addition of the different deoxynucleotide triphosphates. The pyrophosphate released from an incorporated NTP is converted into ATP, which in turn triggers the emission of light from the luciferin � luciferase system. Excess nucleotide triphosphates are degraded by apyrase. As the sequencing reaction progresses, the DNA strand is extended and the sequence is determined from the measured signal output of light upon nucleotide incorporation. The reactions are performed in a 96-well plate in an automated device (PSQ-96, Pyrosequencing, Inc. Uppsala, Sweden). The resulting sequence trace is analyzed automatically by the Pyrosequencing software.
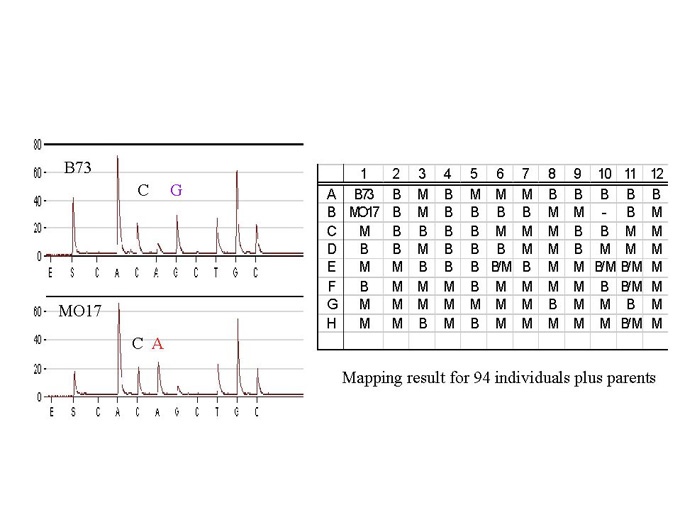
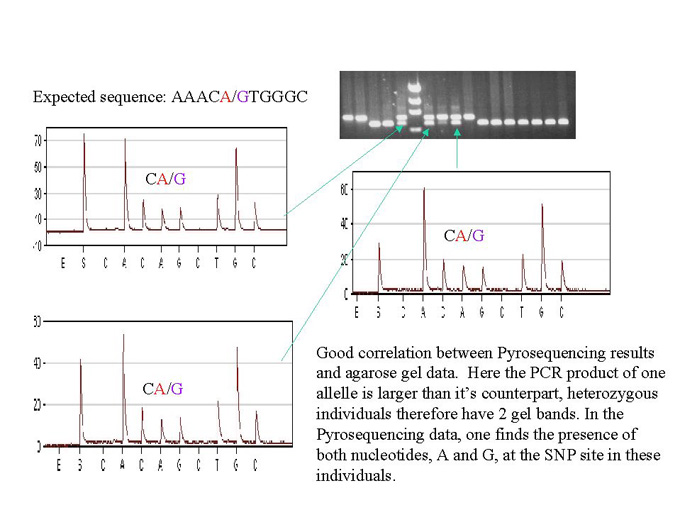
To place an EST on the genetic map, we first identified polymorphisms between the two mapping parents within the EST sequence. This is done by standard dideoxy- terminator fluorescent sequencing of the PCR amplified segments of genomic DNA corresponding to that EST. Sequencing of the 3�-UTR segment of the EST clone increases the likelihood of finding multiple SNPs. SNP frequencies of 1 polymorphism in 60 nt on average are found in a diverse set of maize germplasm (A.Ching, unpublished data). Next, a sequencing primer close to the identified SNP site(s) is prepared. The locus is then PCR amplified from the genomic DNA of the mapping population, this time using one biotinylated and one standard PCR primer. The PCR product is then bound to streptavidin-coated paramagnetic beads, denatured by sodium hydroxide, and a magnetic tool is used to capture the biotinylated template strand for sequencing. Several bases, including the SNP site, are sequenced in the Pyrosequencing machine, using the above-mentioned sequencing primer and the bead-bound template. EST mapping using this approach was tested by mapping the sucrose synthase locus (sh1) using the SX19 mapping population (Lee M et al., Maize Genetics Conference Abstracts 40). The expected sequence read is AAACA/GTGGGC, (SNP position at 4739 GenBank accession X02382, A in B73 and G in MO17, Fig.1). In a heterozygous individual, a half-height peak of both A and G is found. In the case of sh1, the genotypes obtained from Pyrosequencing can be further verified by separating the PCR products on an agarose gel. This is because the PCR product of one of the alleles is larger than its counterpart, due to the presence of a 142 bp insertion in B73 (position 4080 in GenBank accession X02382). A heterozygous individual will therefore have two bands per lane (Fig 2). The genotyping scores were placed on the genetic map of the SX19 population using MapMaker software (Lander, E. and Green, P., PNAS 84:2363, 1987). As expected, the sucrose synthetase locus co-segregated with sh1. We have since placed a number of other EST loci on the SX-19 map using Pyrosequencing genotyping of SNPs, with a success rate of >95% per locus. Failures were due to a poor PCR, and could be rectified by a more careful quantification of the amount of DNA per well before PCR amplification.
Return to the MNL 76 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}
{kind=link}