The role of TnpA and TnpD in transposition of Spm
--Ramesh Raina and Nina Fedoroff
The transcriptional and transpositional activity of the Suppressor-mutator (Spm) transposable element is subject to epigenetic regulation. There are several distinct epigenetic forms of the Spm element which differ in the extent of methylation at the 5'-end of the element. Active elements are unmethylated and inactive elements are methylated at the 5'-end. The 5'-end of the element consists of two regions: the 0.2 kb upstream control region (UCR) which is the element's promoter (Raina et al., Proc. Natl. Acad. Sci. USA 90:6355, 1993) and the 0.35 kb downstream control region (DCR), which is a G+C-rich untranslated leader sequence. The element-encoded proteins TnpA and TnpD are necessary and sufficient for transposition of Spm in tobacco cells (Masson et al., Plant Cell 3:73, 1991). TnpA is a DNA-binding protein (Gierl et al., EMBO J. 7:4045, 1988) and there are multiple copies of its 12-bp binding site located at the element's 5'- and 3'- ends. TnpA affects the epigenetic state of the Spm element by activating the methylated inactive Spm promoter (Schlappi et al., Cell 77:427, 1994). However, the role of TnpA and TnpD in the transposition of Spm is not understood.
The main impediment to studying TnpA and TnpD has been the difficulty of over-expressing these proteins in E. coli. As a consequence, earlier binding studies used either in vitro translated protein or a DNA-protein complex precipitated with anti-TnpA antibodies (Gierl et al., EMBO J. 7:4045, 1988; Trentmann et al., Mol. Gen. Genet. 238: 210, 1993). Here we describe successful production of soluble TnpA from E. coli. TnpA was cloned in the pFLAG MAC expression vector (IBI E. coli FLAG expression system) such that the FLAG peptide at the N-terminus and TnpA are expressed as a fusion protein. TnpA expression in E. coli cells was induced with IPTG. When induction conditions were optimized, more than 50% of the fusion protein was recovered in the soluble fraction (0.1mM IPTG, 25 C incubation temperature at 150 rpm). The fusion protein was then purified by binding to an antibody affinity column containing anti-FLAG antibodies and eluting with FLAG peptide. TnpA purified by this procedure had less than 10% contaminating E. coli proteins and was used directly for DNA binding studies.
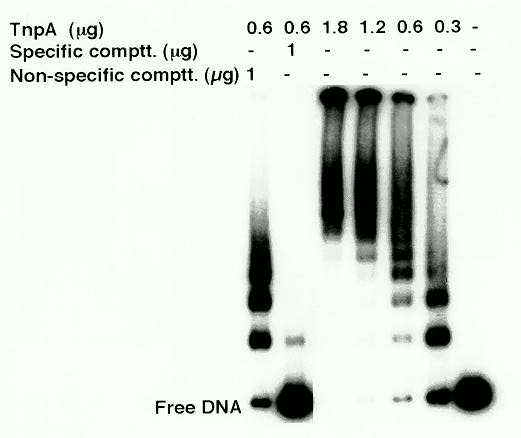
TnpA-DNA binding studies were done using both an oligonucleotide containing two TnpA binding motifs in a tail-to-tail orientation (shown to have the highest binding affinity for TnpA; Trentmann et al., Mol. Gen. Genet. 238: 210, 1993), and the complete UCR. Binding was detected by band-mobility shift experiments. TnpA bound to oligonucleotides containing the TnpA binding motifs and the UCR, but not to the DCR sequence of the promoter (control). When TnpA was bound to the UCR, the mobility of the labeled DNA fragments decreased with increasing protein concentration, forming multiple bands of progressively lower mobility (Fig. 1). These observations suggest that as the amount of TnpA increases, binding sites are occupied progressively until all the sites are filled. The formation of very large complexes that fail to enter the gel suggests higher-order protein-protein interactions between TnpA-DNA complexes.
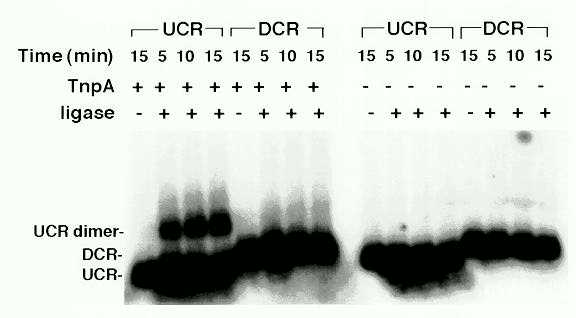
We further investigated the ability of the TnpA to form higher-order DNA-protein complexes by protein-protein interactions. For this purpose we tested the ability of TnpA to accelerate the formation of DNA multimers in the presence of ligase as described by Miron et al. (EMBO J. 11:1205, 1992). TnpA stimulated formation of UCR dimers but not DCR dimers (Fig. 2). Since TnpA binds to the UCR, but not to the DCR, these observations suggest that TnpA mediates the formation of higher-order complexes by interaction of protein bound to different DNA molecules. Because there are TnpA binding sites at both element ends, the higher order interactions of TnpA molecules bound to each element end may serve to bring element ends together during transposition.
We next assessed the ability of TnpD to interact with the ends of the
element and TnpA. Since we have not yet succeeded in expressing TnpD in
E.
coli, we used extracts of tobacco cell lines
over-expressing TnpD cDNA from a 35S promoter as a source of
TnpD, while TnpA was prepared as above. We used oligonucleotides with two
TnpA binding motifs in a tail-to-tail orientation in a mobility shift assay.
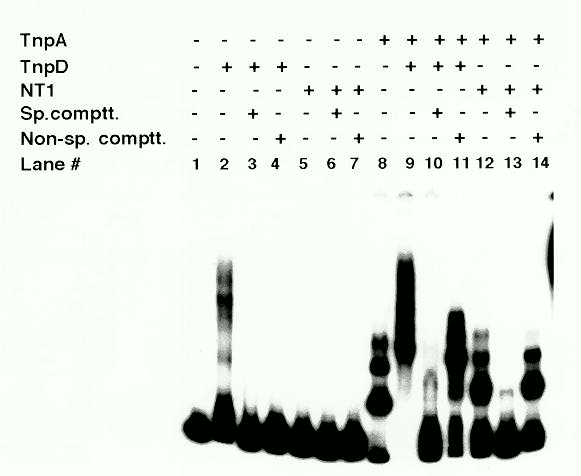
Extracts of the tobacco cells expressing TnpD retarded the mobility of
the test oligonucleotide, while extracts of control NT1 cells did not (Fig
3., lanes 2 and 5). However, the mobility shift was abolished by both specific
and non-specific DNA competitors, indicating that TnpD binds DNA non-specifically
(lanes 3 and 4). As previously reported (Trentmann et al., Mol. Gen. Genet.
238: 210, 1993), TnpA retards the mobility of an oligonucleotide containing
the double TnpA binding sites (lane 8). Addition of TnpD-containing extracts
results in a supershift of the DNA-TnpA complex (lane 9). The supershift
persists in the presence of non-specific competitor DNA (lane 11). Addition
of specific competitor completely abolishes the shift, indicating that
the supershift is due to the interaction of the TnpD with TnpA and not
with the DNA (lane 10). Interaction of TnpD with TnpA is further revealed
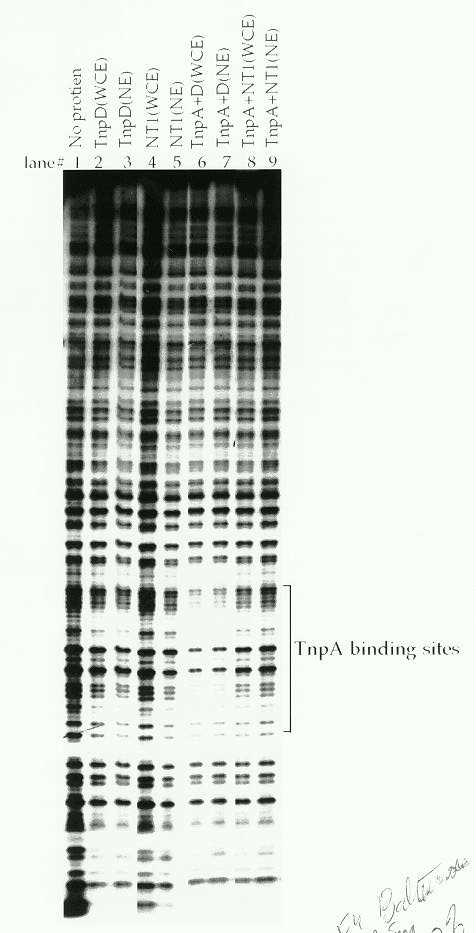
by a DNaseI footprint analysis. As reported in earlier studies (Gierl et
al., EMBO 7:4045, 1988), TnpA alone does not protect its binding sites
from DNaseI digestion (data not shown). However, a DNaseI footprint was
obtained when the oligonucleotide was pre-incubated with both TnpA and
tobacco whole cell or nuclear extracts containing TnpD (Fig 4., lanes 6
and 7). No footprint was observed either with TnpD extracts alone, or when
control NT1 cell or nuclear extracts were used either alone or with TnpA
(lanes 2-5, 8 and 9). Because the TnpA binding site is protected from DNaseI
digestion, we infer that the footprint is due to the formation of a DNA-TnpA-TnpD
complex. The fact that the footprint is obtained with TnpA and TnpD, but
not TnpA alone suggests either that TnpD stabilizes the DNA-TnpA complex
or that the binding of the larger protein to the DNA-TnpA complex restricts
access of DNaseI to this region in some way.
Thus we have shown that TnpA binds to the end of the element, leading to increased occupation of binding sites as a function of TnpA concentration. We have also provided evidence that there are higher-order protein-protein interactions among the TnpA molecules bound to the DNA, which may be responsible for bringing the ends of the element together during transposition. Furthermore we have shown that TnpD is probably not itself a specific DNA-binding protein, but interacts with TnpA to form a DNA-TnpA-TnpD complex. Our current hypothesis is that TnpD is an endonuclease and that its cleavage site is determined at least in part by its interaction with TnpA.
Figure 1: DNA-TnpA complexes analyzed in a mobility shift assay. An 0.2 kb UCR fragment was labeled with 32P-dCTP using Klenow enzyme. Typically 0.1-0.5 ng of labeled DNA was mixed with different amounts of purified TnpA in binding buffer [10% glycerol, 150 mM NaCl, 1mM b-mercaptoethanol, 1mM EDTA, 2 mg poly(dl-dC), 300 mg/ml BSA] in 20 ml reaction volume and incubated at 25 C. The reaction mix was fractionated on a 4% high-ionic-strength polyacrylamide gel (Ausubel et al., Current Protocols in Molecular Biology, 1989).
Figure 2: Ligation-mediated dimerization of blunt-ended DNA fragments. UCR (0.2 kb) and DCR (0.35 kb) fragments were labeled as described in Figure 1. The binding reactions were carried out as described in Figure 1, except that the binding buffer was supplemented with 5 mM ATP, 20 mM DTT, 4% polyvinyl alcohol at 25C for 20 min. 1 mg TnpA was used in the binding reaction where indicated. A 10 ml aliquot was withdrawn from the binding reaction and 4 units of T4 DNA ligase were added to the remaining portion. 10 ml aliquots were withdrawn at 5 min intervals and the reaction was stopped by adding Proteinase K. The entire sample was fractionated on a 1% agarose gel.
Figure 3: DNA-protein complexes analyzed in a mobility shift assay. An 0.15 kb fragment carrying two TnpA binding motifs in a tail-to-tail orientation was labeled as described in Figure 1 and used in the mobility shift assay. Extracts of tobacco control NT1 cells and TnpD-expressing cells were prepared by a modification of a previously described method (Forter et al., In: Methods in Arabidopsis Research, eds. Koncz et al., 378, 1993) and the reactions were performed as described in Figure 1, with the modification that 10 mg of cell extract was added where indicated and 4 mg of poly (dI-dC) was used in the binding reaction when cell extract was added.
Figure
4: DNaseI footprint analysis following the binding of TnpA and TnpD
to an oligonucleotide containing a TnpA binding site. DNA was labeled at
one end as described in Figure 1, binding reactions were performed as described
in Figure 3, and DNaseI digestion was carried out as described by Ausubel
et al. (1989) in Current Protocols in Molecular Biology. Where indicated,
10 mg of whole cell extract (WCE) or 5 mg nuclear extract (NE) was used
in the assay.
Return to the MNL 69 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}
{kind=link}
{kind=link}
{kind=link}