A composite map of expressed sequences, based on four individual
maps.
--Mathilde Causse, Catherine Damerval, Alexandrine Maurice, Alain Charcosset,
Sylvain Santoni and Dominique de Vienne
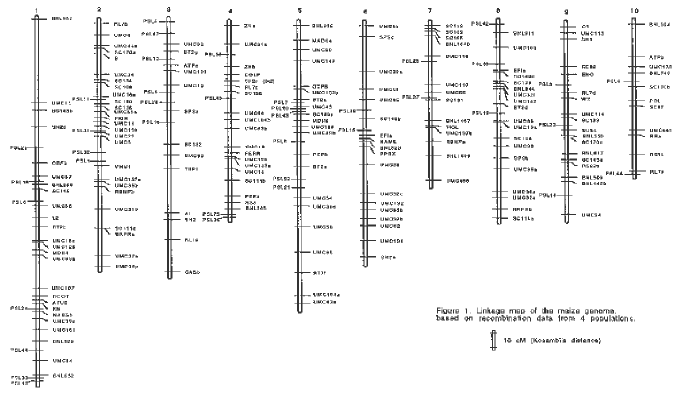
Candidate gene approach is a straightforward way to identify QTLs and to increase the efficiency of marker assisted selection. For this purpose, we are constructing a maize genetic map mainly based on expressed sequences. Our map relies on the data from 4 segregating populations. Three recombinant inbred line (RIL) populations were derived from the three possible crosses between 3 inbred lines, an early flint (F2), an Iodent (coded Io) and an early dent line (F252). A map derived from an independent F2 progeny from the cross Io x F2 was also used. Each progeny contains beween 100 and 150 genotypes, and a total of more than 500 individuals were genotyped. With such sample size, a good confidence in gene order is expected. Around 75% of the tested probes appeared polymorphic in each cross (among more than 200 probes). Four sources of markers were used, and the present map is composed of: - 60 loci corresponding to known function genes obtained from laboratories which cloned them (Table1); - 39 loci (coded PSL) controlling position shifts of proteins revealed by two-dimensional electrophoresis in our laboratory (submitted); - 27 loci (coded SC) of sequenced cDNA for which no homology was found in gene banks, kindly provided by C. Baysdorfer (California State Univ.); - 98 loci of anonymous probes (coded umc and bnl) of the maize core map (Gardiner et al., Genetics 134:917-930, 1993). These markers were useful to integrate our map with the other maize maps.
Mapping cDNA revealed some problems, among which is the high frequency of multiple copy probes. Among the mutiple bands, it is rare that more than one locus per progeny could be mapped. Working with 4 populations and 2 restriction enzymes sometimes allowed maping of a higher number of loci. Depending on the region of the gene used (3' end versus 5' end) we could also reveal different patterns and map additional loci for the Sh2 gene. The development of locus specific probes would be necessary for many known function cDNAs. No specific organization was deduced, except the duplications already mentioned by Helentjaris et al. (Genetics 118:356-363, 1988). Depending on the population, segregation distortions concerned between 4% and 12% of the probes (p<0.01)
Table 1. Chromosomal location of known function genes mapped on the
composite map. Probes whose function has been deduced from sequence homology
are indicated with an asterisk. The percentage of homology is in parenthesis,
with the corresponding organism (ZM: maize; P: other plant; A: animal;
Y: yeast; B: bactery).
| Code | Function | Chromosome |
| A1 | A1, anthocyanin metabolism | 3 |
| ATP* | ATP/ADP translocator (100% ZM) | 3,1 |
| ATUB1 | a tubulin 1 | 1 |
| B | B, anthocyanin metabolism regulator | 2 |
| BRPR* | brain specific 14-3-3 protein (70% A) | 2,8 |
| BT2 | brittle2 (ADPG pyrophosphorylase, endosperm) | 6 |
| C1 | anthocyanin metabolism | 9 |
| CAB* | chlorophyll a/b binding protein (99% ZM) | 3 |
| COLP | cold induced protein | 4 |
| EFI* | elongation factor I a (100% P) | 6,8 |
| ENO* | enolase (99% ZM) | 9 |
| FERR | ferritin | 4 |
| GTPB* | GTP binding protein (79% A) | 5 |
| KN | knotted, transcription factor | 1 |
| L2 | ADPG pyrophosphorylase, leaf | 1 |
| MADS* | MADS box (62% P) | 1,5 |
| MDH* | malate dehydrogenase (68% B) | 1,5 |
| NAME* | NADP malic enzyme (100% ZM) | 6 |
| OBF1 | OCSBF-1, transcription factor | 1 |
| PEPC | phosphoenol pyruvate carboxylase | 4,5 |
| PKIN | protein kinase | 2 |
| POL | pollen specific cDNA | 10 |
| PPDK* | pyruvate phosphate dikinase (100% ZM) | 6 |
| RBNP* | 31 KD ribonucleic protein (60% P) | 2,7 |
| RL19* | ribosomal protein L19 (71% A) | 3 |
| RL7 | ribosomal protein L7 | 2,4,10 |
| ROOT | root specific cDNA | 1 |
| RS | R-S, anthocyanin metabolism | 10 |
| RS11 | ribosomal protein S11 | 10 |
| RS22* | ribosomal protein S22 (76% A) | 9 |
| RS8* | ribosomal protein S8 (70% A) | 4 |
| SH1 | shrunken1 (sucrose synthase) | 9 |
| SH2 | shrunken2 (ADPG pyrophosphorylase, albumen) 3' end | 3 |
| SH2 | shrunken2 (ADPG pyrophosphorylase, albumen) 5' end | 1,4 |
| SPS | sucrose phosphate synthase | 3,6,8 |
| SUS1 | SuS1 (sucrose synthase) | 9 |
| THPI* | thiol protease inhibitor (73% P) | 3 |
| TIOL* | thiol protease (63% P) | 7 |
| WX | waxy | 9 |
| ZN | zein | 4,4 |
Individual maps were first constructed using Mapmaker V3.0 software. A few differences with the core map were detected in the locus position, usually in regard to multiple copy probes. As many loci were common to all maps, we checked for heterogeneity between recombination fractions. The comparison of recombination fractions following the procedure of Beavis et al. (Theor. Appl. Genet. 82:636-644, 1991) procedure revealed: (i) a very good correspondence of the recombination fractions between the F2 and the RIL progeny derived from the same cross; (ii) few significant differences in interval distances between the 3 RIL populations; and (iii) global differences, which can reach 20% of the total map length (when the same subset of loci is mapped). The consistency of probe order over the progenies was confirmed. A composite map has thus been constructed using JoinMap software (Fig. 1). With a total of 233 loci, we approximately cover 90% of the maize genome (when compared with the most recent MNL compilation). The mapping effort is continuing and we would enjoy mapping any known function gene, newly cloned, on our material. The three RIL populations are involved in various QTL location projects (see companion papers), which should lead to a large data set interesting both for maize breeders and geneticists.
Return to the MNL 68 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}