--N. M. Antonelli and J. Stadler
Although methods for introduction of foreign genes directly into intact cells of tissues may eventually become the most desirable method for producing transgenic maize plants, methods for direct transfer into protoplasts remain necessary, especially for preliminary studies of gene structure and regulation (for example, Callis et al. Gene Dev. 1:1183, 1987). We have therefore continued our studies of methods for simple and reliable gene transfer to maize protoplasts.
We report a novel and efficient chemical method for direct gene transfer to maize protoplasts. Freshly isolated protoplasts are treated for 6 or 12h with transfecting DNA and the polycation Polybrene (hexadimethrine bromide: Kawai and Nishizawa, Mol. Cell Biol. 4:1172, 1984). At the end of the incubation period, the transfection mixture is simply diluted by addition of growth medium and the cells are then incubated further for 30h before being assayed for transient gene expression. This gentle method involves little handling of the cells and no physical disruption of the membrane, a feature of both electroporation (Fromm et al., Proc. Natl. Acad. Sci. USA 82:5824, 1985) and microprojectile bombardment (Klein et al., Proc. Natl. Acad. Sci. USA 85:4305, 1988) procedures for transformation of maize cells. And, probably as a consequence of the gentleness of this treatment, we do not detect loss of viability beyond that seen in untreated protoplast control populations by 24h after the transfection treatment (Table 1).
Table 1. Viability after transfection treatments.
|
|
||||
| 0 | 6 | 12 | 24 | |
| Control | 92 | 82(a) | 85 | 71 |
| Polybreneb | 92 | 98 | 70 | 70 |
| Electroporationc | 92 | 59 | 54 | 52 |
aThese values indicate the percentage of viable cells relative
to control at time 0. Each data point is derived from counts of 300 cells
in 3 replicate samples. Viability is ascertained by staining with fluorescein
diacetate.
b2x106 protoplasts were combined with 30ug polybrene and
20ug DNA.
c2x106 protoplasts were transformed with 20ug DNA at 150V
and 800u F for 12.0m sec.
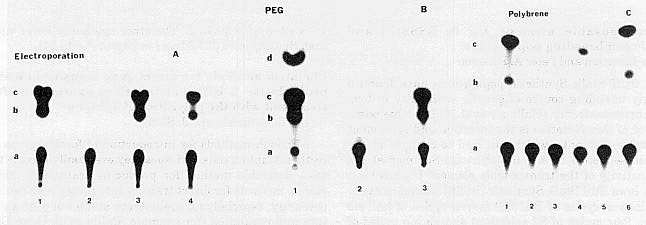
Although it is difficult to make accurate comparisons of the efficiency of direct gene transfer by electroporation (Fromm et al., Nature 319:719, 1986), polyethylene glycol (PEG: Antonelli et al., MNL 62:7, 1988), and Polybrene methods, representative experiments are shown in Fig. 1A, 1B, and 1C. In each case, protoplasts were isolated as described and transformed with 20 to 50ug plasmid DNA, pCaMVI1CN (MNL 62:7, 1988). Assays for transient chloramphenicol acetyl transferase activity were performed 30 to 40h after CAT gene transfer. In all instances (Fig. 1A lane 2; 1B lane 2; lC lane 2) treated control protoplasts without DNA showed no acetylation of 14C chloramphenicol. However, electroporation of 25ug pCaMVI1CN DNA (Fig 1A lane 4), PEG treatment with 50ug (Fig. 1B lane 3), and Polybrene treatment with 20ug of the same DNA (Fig. 1C lane 6) each effect direct gene transfer. In each of these instances, respectively, 6.4%, 37%, and 22% conversion of 14C chloramphenicol to acetylated products was obtained. With all three direct gene transfer treatments we have obtained in different experiments 60% to 95% chloramphenicol acetylation.
Fig. 1. Chloramphenicol-acetyl transferase activity in BMS-M protoplasts transformed with plasmid DNA pCaMVI1CN by electroporation (A), by polyethylene glycol (PEG) (B), or by Polybrene (C). CAT activities were determined by incubating heat-treated lysates with 4mM acetylCoA and 0.1uCi of 14 [C] chloramphenicol. The reaction products were separated on silica gel TLC plates and detected by autoradiography. Reaction products: (a) chloramphenicol; (b) 1-acetylchloramphenicol; (c) 3-acetylchloramphenicol; (d) 1,3-diacetylchloramphenicol. Lanes A: 1, bacterial standard; 2, electroporated control cells without DNA; 3, electroporation with 50ug of pCaMVI1CN; 4, as lane 3 but with 25ug of the same DNA. Lanes B: 1, bacterial standard; 2, PEG-treated control protoplasts without DNA; 3, PEG-treated protoplasts with 50ug of pCaMVI1CN. Lanes C: 1, bacterial standard; 2, Polybrene-treated protoplasts without DNA; 3, control cells with 20ug of pCaMVI1CN without Polybrene; 4, 3x106 protoplasts treated with Polybrene and 20ug pCaMVI1CN, diluted after 6h; 5, 2x106 Polybrene-treated protoplasts with 20ug of the same DNA, but diluted after 18h; 6, 2x106 Polybrene-treated protoplasts with 20ug pCaMVI1CN, diluted after 6h.
Transfection methods using polycations to aid DNA transfer to plant cells are new, although similar techniques have been used for several years in transfection of animal cells (Chaney et al., Somatic Cell and Mol. Genet. 12:237, 1986). Polycations probably enhance adsorption of the transfecting DNA to plasma membrane by interacting with the negative charges of both the DNA molecules and the membrane surface. The adsorption of the extracellular DNA may be followed by endocytosis. When Polybrene was first successfully used in animal transfection it was necessary to follow the polycation treatment with dimethyl sulfoxide (DMSO) treatment to achieve gene transfer. In our initial experiments with the Polybrene/DMSO procedure, we found that the recommended DMSO concentrations (30% v/v) caused protoplast lysis and that Polybrene treatment alone, without DMSO permeabilization, was sufficient to obtain efficient transfection as measured by the transient expression of CAT genes.
The detailed procedure is as follows:
2. For each experiment prepare a fresh Polybrene (Aldrich) stock solution (10mg/ml in phosphate buffered saline, pH 7.0). This is an extremely hygroscopic chemical and the manufacturer's safety instructions must be rigorously applied. The stock solution is then diluted to yield a final concentration of 30ug Polybrene in 0.1 ml MS2D8M.
3. The desired concentration of transfecting DNA is suspended in 0.4ml MS2D8M.
4. Mix the 0.1ml (30ug) Polybrene solution with the resuspended protoplasts and transfer to a 60 mm Petri dish.
5. Immediately add (dropwise) the 0.4ml DNA suspension. The protoplast/Polybrene/DNA mixture (total volume 1.0ml) is rotated gently (25rpm) on a gyrotary shaker for 15 min and then incubated (stationary) at 28 C for 6h.
6. After the 6h incubation, dilute the above mixture with 4.0ml MS2D8M, seal the Petri dish, and follow procedures for assaying transient gene expression or for selection of stable transfectants.
Return to the MNL 63 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}