In order to exploit molecular cloning strategies in genetic studies it is often necessary to compare DNA samples obtained from a large number of individuals. Rapid screening methods have been applied to a variety of microorganisms to follow changes in DNA sequence organization and the insertion of foreign DNA. Isolation of plant DNA is complicated by the presence of a tough cell wall and large amounts of polysaccharides, phenolics, and tannins. Most existing large scale (e.g., Kislev and Rubenstein, Plant Physiol. 66:1140-1143, 1980; Murray and Thompson, Nucl. Acids Res. 8:4321-4325, 1980) and microscale (e.g., Zimmer and Newton, In "Maize for Biological Research," ed. W. F. Sheridan, pp. 165-168; Taylor and Powell, Focus 4(3):4-6) plant extraction procedures, therefore, rely on the isolation of nuclei and phenol extraction or preferential precipitation of DNA followed by equilibrium density centrifugation in CsCl. Such complex and cumbersome techniques are inappropriate for application to a large number of samples and small amounts of tissue.
We report here a plant DNA isolation procedure adapted from one commonly used on yeast (Davis et al., Methods in Enzymology 65:404-411, 1980), which requires 1.0 gm or less of plant leaf tissue and does not rely on isolation of nuclei, phenol extraction or CsCl gradient centrifugation. Partially purified DNA suitable for restriction endonuclease digestion can be obtained from twenty or more samples in only a few hours. Each prep (from 1.0 gram of maize tissue) yields approximately 50 micrograms of total cell DNA, enough for at least ten Southern blots.
An important application of this miniprep procedure in the genetic manipulation of maize is that leaf material may be harvested from seedlings at the 2-3 leaf stage without sacrificing the plant. Information obtained by Southern blotting or other genomic analysis is thus available long before the plants are sexually mature, and can be used in planning crosses involving those plants. For example, we have identified two normal Sh1 isoalleles, in addition to the sh1 tester allele, all of which can be distinguished from one another on the basis of BglII restriction site polymorphisms. By Southern blotting of DNA minipreps, genotypic identification can be made before pollinations are performed. Another instance where minipreps can be used to permit early genetic identification is in a cross such as Sh/Sh x sh/sh; minipreps can be used to screen Sh kernels in the F2 and immediately distinguish homozygous and heterozygous individuals without subsequent crosses. Using the same restriction site markers, we have also found the miniprep procedure useful to screen a number of greenhouse-grown seedlings for a particular genotype, thereby relieving the need to maintain the unwanted plants for more than a few weeks.
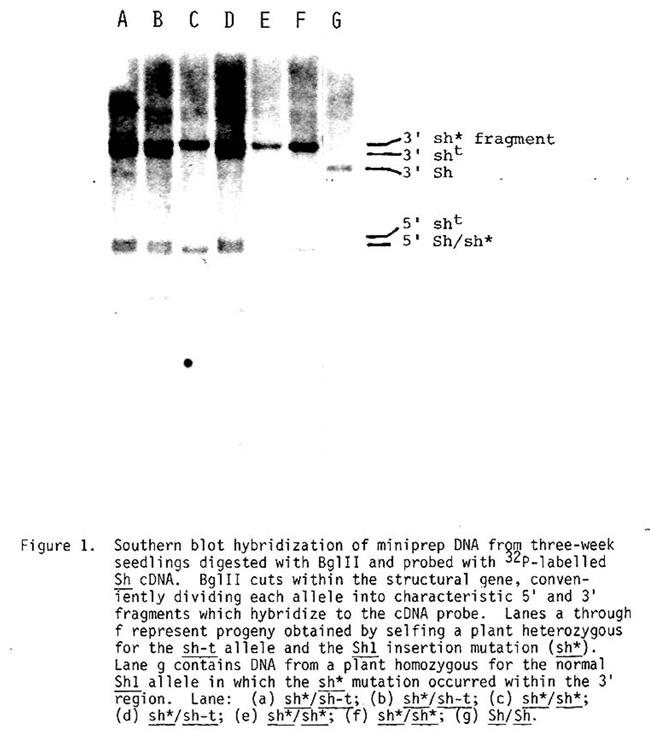
Figure 1 shows a case in which miniprep DNA blots were used to distinguish a standard sh1 tester allele (sh-t) from a newly arisen mutant Sh1 allele (sh*) (Mottinger et al., manuscript in preparation) in the progeny of a selfed sh-t/sh* plant. The DNA was digested with BglII restriction endonuclease, and the blot was probed with the sucrose synthetase cDNA clone obtained from Nina Fedoroff. The sh* mutation is the result of an insertion of a DNA element within the coding sequence (Dellaporta et al., manuscript in preparation) rendering the gene defective. This causes the 3' BglII Sh1 fragment to increase in size by 1.1 kb (compare lane F with lane G). All of these individuals will be shrunken and therefore phenotypically indistinguishable, but the blot clearly shows that the seedlings represented by tracks A, B, and D are heterozygous for the two alleles, while those in tracks C, E, and F are homozygous for, in this case, the insertion mutant.
The degree to which the procedures described above will be generally applicable depends, of course, on the availability of appropriate restriction fragment polymorphisms. Our experience with the Sh1 region, however, indicates that such markers are common in the maize stocks now in use.
Miniprep Procedure
1. Weigh 1 gm of leaf tissue, quick freeze in liquid nitrogen and grind to a powder in a 3" mortar and pestle. Transfer powder with liquid nitrogen into a 30 ml Oak Ridge tube. It is imperative not to let the tissue thaw once frozen until buffer is added and not to cap the tubes while nitrogen is evaporating.
2. Add 15 ml of Extraction Buffer (EB): 100 mM Tris, pH 8; 50 mM EDTA, pH 8; 100 mM NaCl; 1% SDS; 1-0 mM mercaptoethanol. For maximum DNA yields, the cells are further broken by grinding the mixture at a low setting (about 3) with a Polytron (Brinkmann Instruments, Inc.), however, this step can be optional.
3. Incubate tubes at 65 C for 10 min.
4. Add 5.0 ml 5M potassium acetate. Incubate at 0 C for 20 min. Most proteins and polysaccharides are removed as a complex with the insoluble potassium dodecyl sulfate precipitate.
5. Spin tubes at 25,000 x g for 20 min. Pour supernatant through a Miracloth filter (Calbiochem) into a clean 30 ml tube containing 10 ml isopropanol and 1 ml 5M ammonium acetate. Mix and incubate tubes at -20 C for 20 min.
6. Pellet DNA at 20,000 x g for 15 min. Wash pellets with cold 70% ethanol and respin. Gently pour off supernatant and dry pellets by inverting tubes onto paper towels for 5-10 min.
7. Redissolve DNA pellets with 0.7 ml of 50 mM Tris, 10 mM EDTA, pH 8. Transfer the solution to an Eppendorf tube.
8. Add 75 ul 3M sodium acetate and 500 ul isopropanol. Mix well and pellet the clot of DNA for 30 sec in a microfuge. Wash pellet with 70% ethanol, dry, and redissolve in 100 ul 10 mM Tris, 1 mM EDTA, pH 8. Precipitation from 0.3M sodium acetate using relatively small amounts of isopropanol (about 0.6 volumes) has been reported to separate high molecular DNA from polysaccharides (Marmur, J. Mol. Biol. 3:208-218, 1961). The sodium acetate also yields a tight, fibrous precipitate that is easily washed and dried. The DNA will dissolve readily if allowed to rehydrate at 4 C for one hour, followed by light vortexing.
Minipreps can be stored for several months without evidence of degradation and can be cut with a variety of restriction enzymes and ligated without further purification. We find that 7.0 ul of miniprep DNA is sufficient for a single 8 mm lane in an agarose gel which is to be used for filter hybridization with single-copy probes. Heat-treated RNAase must be added to the restriction reaction to digest contaminating RNA in each prep. Hence, a typical reaction would contain the following:
Miniprep DNA 7.0 ul
10X Restriction Buffer 2.5 ul
0.5 mg/ml RNAase 2.0 ul
Eco RI 8 units
dH20 to 25 ul
Digestion is usually complete after 3 hours at 37 C. Occasionally, minipreps are difficult to digest with certain enzymes. This problem can be overcome by adding 5.0 ul of 0.1M spermidine to the entire miniprep before digestion (see Focus 4(3):12, 1982). For lambda genomic library construction, we have found the packaging efficiencies are higher if the minipreps are further purified by CsCl-ethidium bromide gradient centrifugation. This can conveniently be done by pooling 2-5 identical minipreps per gradient.
Stephen L. Dellaporta, Jonathan Wood and James B. Hicks
Return to the MNL 57 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}