We have applied standard techniques of DNA reassociation analysis to determine the organization of the genome of Zea mays. DNA was prepared from seedlings, denatured, reassociated, chromatographed on hydroxylapatite, and the resulting reassociation data analyzed by a sum of squares curve-fitting computer program. The major finding is that the corn nuclear genome contains three kinetic classes of DNA: very rapidly reassociating (low Cot), fast reassociating (mid Cot) and slow reassociating (high Cot) DNA sequences; these three kinetic classes are general features of all eukaryotic DNAs so far examined.
Reassociation experiments were performed with DNA fragments sheared to an average of 1350, 1100, and 500 base pairs; only the data from the 1350 base pair material are presented here. At all fragment lengths, however, computer analysis demonstrates three major kinetic classes; changes in fragment length change the proportion of total DNA in each class due to the interspersion of the kinetic classes in the genome.
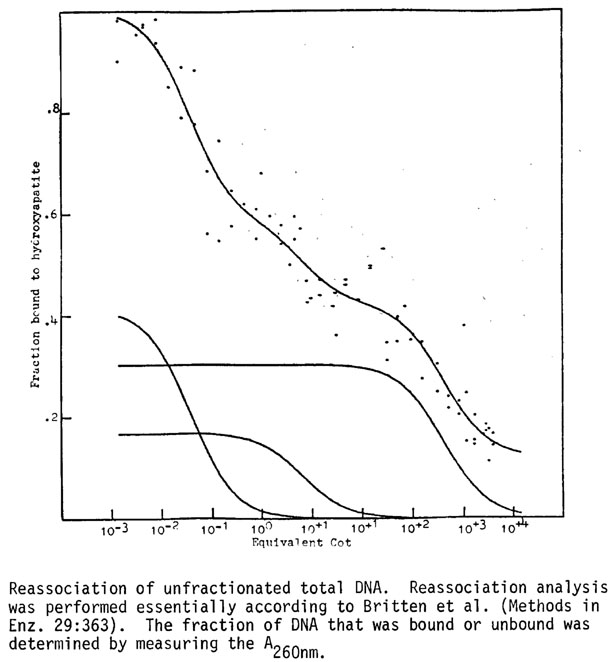
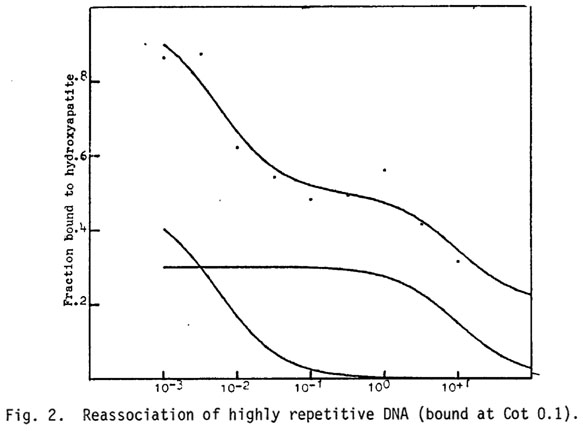
Low Cot, rapidly reassociating DNA: The low Cot DNA appears to be a large portion of the total genome (Figure 1) when whole genomic fragments of 1350 base pairs are analyzed; this is due to the interspersion of this component with mid and high Cot DNA. Minicot data (Figure 2) of this kinetic class, defined as DNA bound to HAP at Cot 0.1, show that this component is composed of several subclasses. About two-thirds of the minicot curve is a component of very low Cot-1/2 (0.0025) indicative of palindromic DNA and extremely repetitive DNA; the remaining one-third of the DNA is a portion of the mid-repetitive class with a Cot1/2 of about 2.88 in this figure. The complexity of the very rapidly reassociating DNA calculated to be about 20% of the genome is 2 x 106 daltons.
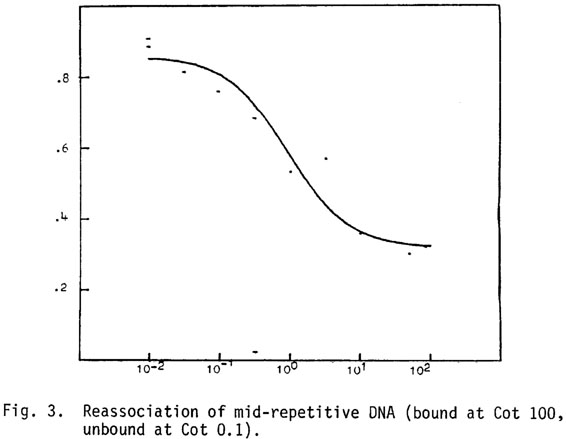
Mid Cot, fast reassociating DNA: When whole genomic DNA is analyzed the middle repetitive component is about 20% of the genome with a Cot1/2 pure of 1.059 and a complexity of 7.06 x 108 daltons. A more direct analysis of the Cot (pure) value is obtained from minicot curves (Figure 3). When mid Cot DNA is purified as DNA reassociated at Cot 100, not reassociated at Cot 0.1, a single component approximately 40% of the genome is found with a Cot1/2 pure of 0.917. Spillover of mid Cot DNA into the very rapidly reassociating component occurs at 1350 base pair fragment length in the whole genome preparations (Figure 1).
High Cot, slow reassociating DNA: The high Cot, presumably unique copy, DNA has a Cot1/2 pure of 1190 which gives a complexity of 7.93 x 1011 daltons; the unique copy DNA is about 30% of the genome. There would be approximately 8 x 105 different gene size sequences of unique copy DNA in the haploid maize genome.
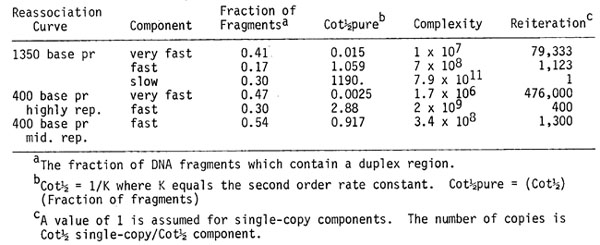
Organization of the genome: As tabulated below, it is possible to calculate the reiteration frequency and number of individual sequences involved in the repetitive components. At 1350 base pair length there are approximately 80,000 copies of each very fast entity; if shorter DNA is analyzed, e.g. 400 base pair DNA, the apparent reiteration of this component increases to 470,000. Estimates of the reiteration of the fast, mid Cot component range from 400-1,300 for each family. The variability in these estimates is due to the relationship between the length of molecules used in the experiment compared to the actual average length of members of each kinetic class. Small changes in fragment length can produce profound changes in the apparent reassociation kinetics.
We have analyzed the average lengths of members of each kinetic class by electron microscopy. EM measurements of S1 nuclease-treated duplexes at mid Cot values give an average of 500 base pairs for the repetitive DNA sequences. The length of the average unique sequences was obtained by reassociating DNA of different fragment lengths to Cot 50. As fragment length increases, the percentage DNA in duplex increases due to single-stranded tails of unique DNA connected to the reassociated repetitive DNA. An estimate of the length of the unique DNA is obtained from the breakpoint in % duplex vs. fragment length; the length of the interspersed unique sequences from this analysis is 2100 base pairs.
Using this information, it is possible to formulate a model for the general features of organization of the genome. At Cot 50 and long fragment length, all the repetitive DNA and two-thirds of the unique DNA are HAP bound. Given the calculated lengths and distribution of kinetic classes, our data indicate that two-thirds of the unique and about one-twentieth of the repetitive-DNA are in a short term interspersion pattern. The remaining one-third unique DNA is probably in a long-term interspersion pattern with large blocks of repetitive DNA.
Preliminary data from melting profiles were also obtained for native and reassociated DNA samples. The native DNA has a Tm of 86.5 in 1x SSC, hyperchromicity of 30%, and a dispersion 2/3 of 13 (H.R. Mahler & Dutton, G. J.M.B. 10:157, 1964). Reassociated DNA to mid-Cot values (Cot 100) has a Tm of 79.5, hyperchromicity of 12%, and a dispersion 2/3 of 18. This indicates that the repetitive duplexes have 6% mismatch, that only 40% of the fragments are in duplex (at 1350 base pairs), and that the heterogeneity is much greater than native DNA. DNA reassociated to higher Cot values (Cot 3000) has a Tm of 85, hyperchromicity of 20% and dispersion 2/3 of 15.5. Thus there is less mismatch at the higher Cot values but still a small percent (1.5%), indicating some of the duplex molecules are not perfectly reassociated. The melts and further data will be published elsewhere.
Sarah Hake and Virginia Walbot
Return to the MNL 53 On-Line Index
Return to the Maize Newsletter Index
Return to the MaizeGDB Homepage
{kind=link}
{kind=link}
{kind=link}
{kind=link}